


患者社区

神经纤维瘤病分三型:1型神经纤维瘤病、2型神经纤维瘤病和神经鞘膜瘤病。2型神经纤维瘤病(先前的中枢神经纤维瘤病或双侧听神经瘤病)首次描述是在1822年的一个患有颅骨、硬脑膜和脑肿瘤的耳聋的患者。52几十年来,2型神经纤维瘤病常常与更常见的1型神经纤维瘤病(即先前的von Recklinhausen病或 周围神经纤维瘤病)相混淆。531920年,2型神经纤维瘤病的遗传性在一个三代患有前庭神经瘤的家系得以描述。54而它的常染色体显性遗传是在1930年一个五代38个患者的家系报道的。551987年,1型神经纤维瘤病和2型神经纤维瘤病通过基因连锁分析而确定为不同的染色体的突变,这两种疾病正式分开。56-58
1型神经纤维瘤病是由位于17q11的NF1肿瘤抑制基因突变引起的,遗传和表现明显不同的常染色体显性遗传的肿瘤综合征。临床表现为多发的咖啡斑、多发的皮肤和皮下神经纤维瘤、丛状神经纤维瘤、雀斑、视觉传导通路的神经胶质瘤、Lisch结节和骨发育不良。58-60随后,确认神经鞘膜瘤病为第三类的遗传性周围神经肿瘤综合症,并确定其由位于22q11的SMARCB1肿瘤抑制基因的突变相关。61临床症状为多发除前庭神经鞘瘤外非真皮内的神经鞘瘤。62
根据2型神经纤维瘤病多种临床表现的研究表明,1.4.63该病的表型表达和自然史在家庭成员间极为相似,但在NF2患者家庭间存在较大差异。目前已被证实,特殊类型的基因型和表现型之间存在相关联系,这有助于阐释家庭内部存在的临床异源性。一般来说,体细胞NF2基因的无义表达或者移码突变产生的去顶蛋白与严重疾病有关,而错义突变和结构内或大的染色体缺失与轻微的疾病相关。64.65尽管1-5位点外显子突变导致相关的疾病比11-15外显子更严重,但剪接位点的突变造成的疾病严重程度仍存在差异。66
这些基因型-表现型效应与死亡率相关风险有关。与携带无义突变或移码突变的NF2患者相比,错义突变的患者的死亡率更低。20虽然突变类型与疾病特征(颅内脑膜瘤和白内障的存在,脊髓肿瘤和周围神经肿瘤的的数目)存在联系,但是特殊肿瘤的行为似乎不依赖于突变类型。6.11.64.67-69
由于新发突变的患者高频率的体细胞镶嵌现象,使2型神经纤维瘤病成为一种不同寻常的遗传性疾病。70-72对于体细胞镶嵌的患者,突变发生在受孕之后,最终形成两个分离的细胞谱系。大多数体细胞镶嵌的患者一部分细胞携带这种突变,但由于外周血中淋巴细胞数量太少而未能检出基因突变,但肿瘤组织可以检测到。临床特征的范围预示了 在镶嵌现象患者的生长发育过程中,当突变发生并占主导地位时,患者往往出现轻微全身症状或局限的病变(例如,单侧的前庭神经鞘瘤伴同侧的肿瘤)。
NF2体细胞镶嵌现象的患者,其仅有部分生殖细胞可能携带这种突变,因此将这个突变遗传传给后代的概率将小于预期的50%(1/2)。可是,从具有镶嵌现象父母那里继承这个突变的儿童将很可能出现比他们父母更严重的病变,因为突变存在于他们所有体细胞。临床评估表明,33%的新发双侧前庭神经鞘瘤的病人和高达60%的单侧前庭神经鞘瘤的病人都存在镶嵌现象。
在患者出现症状前进行基因检测是家庭治疗的重要手段,因为早期诊断能够更好的进行家庭临床护理。当受累患者确定存在某个突变,家庭高危成员的DNA中就有可能也被检测出这个突变基因。携带这个疾病的遗传形式患者,对所有外显子直接进行基因序列分析和探针扩增的联合应用,这个突变的检测敏感度达91-95%。21.73新发NF2的患者(可能有镶嵌现象),对其血样和肿瘤样本进行外显子扫描的分层分析,并结合通过多重连接依赖性探针扩增进行缺失和复制分析,将会检测出80-85%的突变。74
在英国对存在患NF2风险的儿童,进行症状出现前的基因检测,推荐在10岁左右进行。--大多数的医学中心会对在这个年龄的这些孩子实行MRI筛查。在这个年龄段进行基因检测的理由是基于来自许多家庭,MRI研究会显示。而在美国,是依据个别家庭的需要,而对存在风险儿童进行症状出现前的基因检测年龄。当父母认识到孩子携带这种突变,就可以更好的着手监护检测早期的疾病,并让家庭或患者为这个疾病即将出现的相关事件做好充分准备。75
虽然携带种系突变的父母遗传给子女是常染色体显性遗传类型(1/2的风险),但是新发突变的父母遗传给孩子的风险实际上低于常染色体显性遗传类型。血液突变分析阴性和双侧前庭神经鞘瘤的新发NF2患者遗传给后代的几率是1/8。但新发患者只是单侧前庭神经鞘瘤的症状时,风险降为1/12。其发生风险的减小与生殖母细胞种群,其在各种临床状态下突变降低的可能性成正比。72
神经学表现
前庭神经鞘瘤
双侧前庭神经鞘瘤是NF2的特征表现,可见于90-95%的患者。3.4.5散发性的前庭神经鞘瘤大多起源于第Ⅷ对脑神经(前庭蜗神经)前庭部下部,但是NF2并不常见。76-78虽然99%的NF2的前庭神经鞘瘤是良性的,但是因为发生部位,它们仍是死亡的重要原因。79
60%的成年患者和30%以上的儿童患者的主要症状为听力丧失和耳鸣(在病初期常为单侧)。18.21.49-51虽然,有研究回顾性分析了近期确诊为NF2患者的听力结果,80认为患侧在未接受治疗的情况下听力能保持稳定两年左右。但部分患者可表现为进行性听力丧失,与肿瘤大小和生长速度无关。患者双耳听力丧失的几率往往也是不同的。80有研究对表现为前庭神经鞘瘤的NF2患者进行纵向对比,通过Meta分析显示81这些肿瘤生长变量增长比率随着年龄的增大反而减小。
前庭神经鞘瘤检查最佳手段是通过高分辨率增强核磁T1加权成像,可见明显强化。T2加权或液体衰减反转恢复(FLAIR)成像序列用来精确显示瘤周水肿和囊肿。
在手术中, NF2相关的前庭神经鞘瘤常为棕褐色,与散发神经鞘瘤相比为更多分叶的生长模式。53.82其特征性表现为神经鞘瘤与相邻脑神经粘连更紧密,且掺合更多的正常神经束膜。83组织学研究显示了梭形细胞呈混合密度或较疏松密度排列,常常能观察到贝罗凯体(Verocay bodies)、强化的细胞核多型性现象以及透明化的血管。
因NF2导致听力丧失的基因型-表现型的直接联系尚未确定,并且肿瘤的大小和生长速度不能预测听力改变状况;因此,私人诊所医生或各专科医生个人经验在很大程度上影响了NF2前庭神经鞘瘤的治疗。手术全切肿瘤是有效地治疗方法,但是手术治疗的时机仍存在争议,应力求在外科治疗风险与肿瘤的自然发展史找到平衡。小前庭神经鞘瘤(直径小于75px)的早期外科治疗可保存患者30-65%的有用听力和75-92%的面神经的正常功能。7.76该治疗体系目前普遍应用于双侧听力完整的患者。当一侧听力保留成功时,可考虑双侧的肿瘤切除术。即使听力已丧失,早期的肿瘤移除可能有助于保持蜗神经的完整性,从而可通过耳蜗植入术恢复听力成为可能。
但目前总体而言,实际外科治疗NF2--即使是小肿瘤切除手术,在保护听力和面神经的效果上比报道要差得多。7.76这个差异可能归因于:患者在得到专科医院(即报道良好效果的医疗机构)的合理化治疗前,往往接受了地方医院及外科医生的处理造成的,但这往往也是初级医疗水平必须经历的学习过程。84.85因此,传统的治疗策略通常是保守的,2-75px大小的肿瘤患者只有在听力已经丧失或肿瘤快速生长时才被建议进行手术治疗;而当肿瘤增大超过2-75px范围之外时,那些想坚持保留听力的患者应被告知手术风险(如面瘫,后组颅神经损伤)将随着肿瘤的生长而增加。虽然明白这种观望的方法将会导致手术风险的增加(包括面瘫和不可避免的听力丧失),一些病人还是可能会选择等待,直到出现脑干压迫症状或颅内压过高而不得不手术摘除肿瘤。
在不延误治疗的同时为避免手术风险,部分研究机构研究人员观察了立体定向放射技术治疗NF2的前庭神经瘤的潜在效果。小肿瘤(小于75px)和那些没有瘤周水肿或囊变的前庭神经鞘瘤,最有可能对手术起反应。有研究报道通过局部放射治疗74-100%为NF2前庭神经鞘瘤(平均随访54个月),肿瘤能控制在稳定体积,33-57%和92-100%的患者适度听力和面神经正常功能的保留86–89。由于长期随访显示79,90立体定向放射治疗由于需要接受持续的局部照射,并且存在放射后诱发恶性疾病,故其成为首选治疗NF2听神经鞘瘤的方案还有待进一步证实。此外,由于长期放射治疗后引起的瘢痕形成增加了今后手术中保留面神经功能难度,并且影响前庭蜗神经功能和活性,为进一步听力重建造成了困难。
由于双侧前庭神经鞘瘤引起的双侧听力丧失发生率较高,从而带动了听力重建技术的发展。当患者双侧听力丧失但拥有解剖上和生理上完整的蜗神经时,可选择耳蜗植入永久改善听力91,92。当蜗神经生理功能完整性破坏时,听觉脑干植入可能成为其听力重建较好的选择。
关于听觉脑干植入的理想时间仍然有争议。一些中心在进行首次前庭神经鞘瘤切除时就放置听觉脑干植入电极,以备将来使用。而其他中心往往在二次肿瘤手术时才植入听觉脑干植入电极。携带这些植入电极的NF2患者在环境声音和唇读法辅助下可获得有限的帮助,但仅仅至是极少的声音也能明显改善语言理解力。ABI是通过对蜗神经核的刺激获得听力,目前有机构开始对听觉中脑植入刺激下丘进行研究(临近听觉通路的位置)95。
脑膜瘤
脑膜瘤是与NF2相关第二位最常见的肿瘤。颅内脑膜瘤见于45-58%的患者,硬膜内髓外的脊膜瘤出现于约20%的患者。3,4,5,10NF2患者颅内脑膜瘤常常为多发占位,且发病年龄较散发脑膜瘤更早。4,5,49,51在出现脑膜瘤的儿童中20%以上患有NF2,此类患者迫切需要进行全面的临床筛选和长期的随访。51,96脑膜瘤产生的临床症状与其大小和解剖位置相关。虽然大脑凸面脑膜瘤常发展到较大体积才会产生临床症状,但是那些临近重要结构如视神经鞘、颅底及椎管的小肿瘤,早期就会引起很明显的症状。发生颅内脑膜瘤患者相对死亡率比其他NF2患者高2.5倍20。
脑膜瘤可均匀增强,在对比增强的MRIT1加权像最易辨认。MRIT2加权像和FLAIR序列可显示瘤周水肿和肿瘤闹变。硬膜尾征常指强化区域从肿瘤中央沿硬膜扩展强化。脑膜瘤镜下特性征为淡红褐色组织,有完整包膜。所有主要的组织学亚型(乳头型、成纤维细胞型、沙样瘤型和过渡型)均能在NF2中出现,其中成纤维细胞型易变性高于平常。98.99NF2相关脑膜瘤的细胞常增殖活跃,间变型和不典型的出现率高于散发脑膜瘤。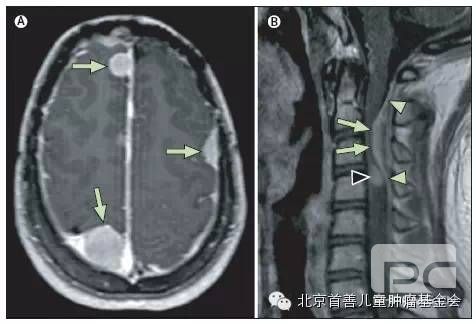
大多数大脑半球和脊椎管的脑脊膜瘤可安全的完整切除。对起源于视神经鞘和颅底的脑膜瘤,由于解剖位位置深在,如手术完整切除则可能引起严重的神经系统并发症。99.100对于手术受限区域残留的脑膜瘤,部分个案报道采用了立体定放射辅助治疗101,102。其中一项研究显示了103立体定放疗NF2脑膜瘤,控制肿瘤瘤发展的同时患者带瘤生存率为86%。但是这项研究长期随访未提及放射治疗诱发肿瘤恶性变和其他相关肿瘤发展情况。104即使目前研究已获得这些可靠的结果,但临床医师必须权衡对进行性发展导致高致残率NF2采用放射治疗引起的潜在损害和外科手术在治疗高危脑膜瘤存在的风险。
脊髓室管膜瘤
2型神经纤维瘤病的髓内肿瘤75%以上为室管膜瘤11,10,105,106。它们见于18-53%的NF2患者,但是少于20%的室管膜瘤会引起临床症状。10-13这些髓内肿瘤的发生常见于发生无义突变和移码突变的患者。10
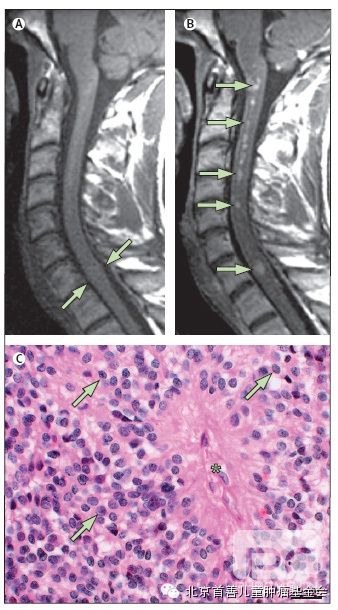
脊髓室管膜瘤产生的临床症状取决于它们沿脊髓长轴的大小和位置。髓内病变患者的典型临床症状为平卧背部疼痛(56%),无力(28%),感觉障碍(16%)。107–111脊髓室管膜瘤在不强化的MRIT1-加权像上是等信号或稍高信号,给药后均匀强化,沿脊髓中央管的多发变在对比增强的MRI显示为类似串珠样表现。10-13室管膜瘤在T2加权像是高信号,并可观察到肿瘤上极和下极的含铁血黄素帽。髓内室管膜瘤镜下为紫红-黄褐色组织。组织病理学分级为WHO II级,典型HE染色表现为中度圆形或卵圆形细胞核的肿瘤细胞,偶见血管周围假菊形团和室管膜的玫瑰花形排列(图5)。手术可有效治愈引起症状的脊髓室管膜瘤,而无症状得室管膜瘤多采取持续观察。由于多数室管膜瘤常因静止或无症状而存在许多年,而手术存在相关风险,故尽可能避免了手术治疗。术后最好的神经功能预后就是同术前状态,详细的神经系统监测确定是否处理早期症状和体征有助于确定最佳手术时间。107,108,111由于大多数肿瘤为WHOII级,镜下全切后常不需要辅助其他治疗。
周围神经系统疾病
大多数NF2患者在会发生周围神经病。虽然很多病例可归结为肿瘤皮内生长或压迫了神经,但是一些病例与肿瘤无关。研究表明3,4,14,66%以上的NF2患者发生神经功能障碍与肿瘤压迫无关。
NF2非肿瘤相关性周围神经病变的病例报道提高了对此类疾病的认识,114-121这些患者常表现为:局限性的肌萎缩、对称性末梢感觉运动神经病变、多发单神经病变等。发病年龄7-41岁,症状持续时间为3个月-50年不等。周围神经病变(如面神经麻痹、足下垂)可早于肿瘤(如前庭神经鞘瘤、脊髓肿瘤)出现,在确诊NF2之前可作为警戒性的发现。51
周围神经病变不能用脑和脊髓MRI检查发现的肿瘤来解释,需要应用电生理技术检查,包括神经传导和针肌电图描记。核磁-神经影像技术可用于对未明确的周围神经肿瘤或导致局部神经病变的非新生肿物的诊断。122
NF2周围神经病发生的可能机制:由于沿邻近周围神经长轴生长的多发微小瘤累积压迫该神经,引起神经鞘肥厚病变(施万细胞增生伴轴突缠结,但无明显肿瘤形成),病态细胞局部产生毒性或代谢终产物反应,或者是归因于merlin蛋白单倍剂量不足引起施万细胞的机能紊乱。14,123
周围神经病变的患者的周围神经的组织病理始往往显示有髓鞘或无髓鞘纤维的缺失以及非正常的施万细胞增殖,伴有或不伴洋葱皮样改变。14,114–120,123周围神经病变的治疗与NF2患者其他症状的治疗不同,主要原则为对症处理,包括神经性疼痛的内科治疗(如加巴喷丁,普瑞巴林)和预防与缓解治疗。
其他神经系统表现
其他神经系统病变最常见的是源于神经鞘膜的肿瘤-神经鞘瘤,其沿非前庭颅内神经(III-VII,IX-XII)、脊神经和周围神经长轴生长。51%以上的NF2患者患有(除前庭神经鞘瘤以外)颅神经鞘瘤,这些神经鞘瘤最常见于脑神经Ⅲ(眼球运动),Ⅴ(三叉神经)和Ⅶ(面神经)(表1)。5.7-9虽然后组颅神经肿瘤的发生常常少于前组颅神经,但是后组颅神经肿瘤往往与临床常见症状相关。神经鞘瘤似乎不在Ⅰ(嗅神经)和Ⅱ(视神经)上生长,可能是因为它们是中枢神经系统的衍生物,没有施万细胞包绕。
脊神经根的神经鞘瘤常是多发的,几乎占髓外肿瘤的90%。10.12其中30%的患者因神经鞘瘤出现了临床症状而切除肿瘤(表1)。3.10.12神经鞘瘤可沿着周围神经走行发生于任何部位,导致间断的周围神经病。43%-48%的NF2患者发现有呈结节状皮下神经鞘瘤,其常引起神经并对压力敏感。3.19神经纤维瘤在NF2所有肿瘤中罕见,其主要通过组织学检查与神经鞘瘤相鉴别,表现为施万细胞松散、随机排列,纤维母细胞内有胶原-粘液样的基质。引起症状周围神经病变主要通过外科切除治疗。
脊髓内偶见星形细胞瘤(弥漫型和毛细胞型),髓内的神经鞘瘤已在NF2中有过报道。10-13,105,106,124
典型的弥漫性星形细胞瘤是边界不清的T2加权像高信号不强化占位,与室管膜瘤相比,在脊髓内占据了更明显的位置。毛细胞性星形细胞瘤和髓内神经鞘瘤在T1加权像上均呈现强化,通过神经影像学鉴别困难。T2加权像病变周围呈高信号的髓内占位,多提示是神经鞘瘤。125此类症状性病变主要治疗手段为手术切除。126辅助的放射疗法可用来治疗残留的弥散星形细胞瘤,在个别病例中能患者提高生存率,但是这些发现在NF2的适用性仍不确定。127
良性的颅内钙化沉着物(CT上)常可见于脉络丛、小脑及大脑实质内。128尸检的研究已证实了在大脑皮质和基底神经节存在小的发育不良的神经胶质病灶集中点。这些神经胶质错构(微小错构瘤)似乎并不是癌前病变。129皮层多发的脑膜血管斑板样增殖即脑膜血管瘤病,需尸体解剖时鉴别,但与NF2的癫痫无关。130
视觉的表现
晶状体浑浊是重要的诊断特征。60-80%的NF2患者患有白内障(表1)。3-5.15只有白内障见于小于50岁的患者,可认为是此病的特征,包括浑浊位于晶状体囊膜下后方、囊膜内及晶状体周围的皮层区)。131.132体积小的和位于皮层边缘周围的浑浊需要在最大散瞳后进行仔细的眼科检查。在10-25%的患者视力受白内障影响而需要摘除。
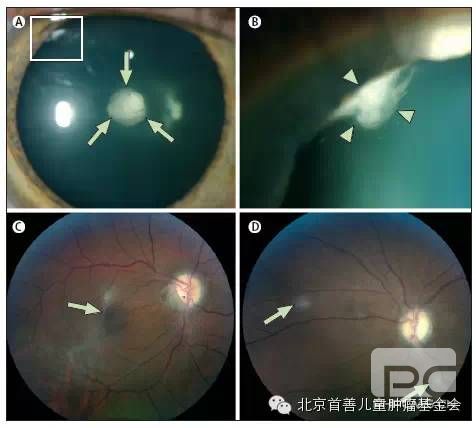
其他视觉的表现为视网膜外膜和视网膜的错构瘤。134.135视网膜外膜是以突起的灰色边缘为界,透明的、半透明的或灰白色的膜。即使是严重临床表现的NF2患者,80%的可见视网膜外膜,这些膜通常不是引起视觉丧失的原因。136视网膜错构瘤(6-22%的患者)表现是轻微隆起的包块,最常见于黄斑,常引起视力下降。4.5.16眼底镜检查时,特征性表现为增强的色素沉着,因增厚的厚度、灰白色的视网膜和视网膜外层组织的数量而不同。137
皮肤表现
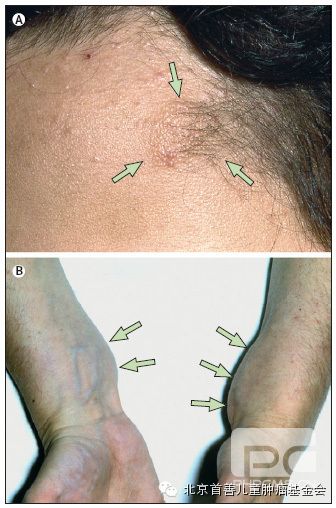
皮肤肿瘤出现于59-68%此疾患者,包括皮肤斑块、皮下肿瘤和皮内肿瘤。3.4.19在严重的患者出现频率有所增高,且常是多发(皮肤肿瘤数量的平均值7.1)。3.4.19其好发部位尚未得到证实。虽然大多数的皮肤肿瘤是神经鞘瘤,但是也有神经纤维瘤或混合型肿瘤通过组织学证实发现。皮肤斑块是一片局限、轻微隆起、粗糙不平的区域,典型病变小于2厘米且表现为色素沉着过度和多毛症。3.19.138此症存在于41-48%的患者。但10岁以下患者往往表现为无毛、光滑且柔软的区域。3.17.19皮下肿瘤沿外周神经走行生长,可见或触及为梭形或结节状隆起,出现于43-48%的患者,常常引起疼痛和压力敏感。3.19在NF2患者中,皮内肿瘤较其他皮肤病变少见,表现为侵及皮肤的、边界清楚的、柔软的紫色病变。牛奶咖啡斑是NF2非特异性表现,可见于33-48%的患者,定义为皮肤上平坦的、色素过度沉着的区域,在NF2中经常是单发的,不明显的。
疾病的进展和生存率
NF2首发症状的平均年龄是20岁,但是临床确诊时间相比平均延误7年。3-5.139虽然该病进展呈现高度差异性,但是绝大多数患者最后出现耳聋,且往往最终需要轮椅的辅助。诊断为颅内脑膜瘤的患者,首发症状出现的年龄越小,疾病越严重,早期的死亡率的风险也相应增加。20在1992年,虽然在一个有150名患者同期组群中,平均存活时间62年,但这些人群中40%以上的患者预期在50岁死亡。3从那时起,由于接受专业中心的有效治疗和对镶嵌现象患者轻微临床表现的认识,所以15年平均生存时间可能从诊断时起被延长。
结论
随着对NF2临床特征认识的不断提高,并且联合基因检测和影像学表现,目前已经提高了对患者的早期诊断。而对该病分子生物学发病机制和病变自然进程更深入的探索,以及康复模式不断改进,将共同促进NF2的治疗。
